
월요일
데이터 전처리 1 (Data Cleaning 1)
- Data Cleaning 1
1 2 3 4 5 6 7 8 9 10 11 12
raw_dat = read.csv(file = "Ex_data.csv", header = T, stringsAsFactors = F) head(raw_dat[,1:20]) dim(raw_dat) gr_ind = gl(2, 221) gr_ind dat_mat <- t(as.matrix(raw_dat[,-1])) dim(dat_mat) rownames(dat_mat) <- paste0("s",1:nrow(dat_mat)) colnames(dat_mat) <- raw_dat[,1] head(dat_mat[,1:20])
- csv파일로 저장된 Ex_data.csv파일은 Lung Cancer Microarray data 이다. 특정 유전자의 발현량이 폐암에 영향을 미치는 가에 대해 분석할 때 사용하는 데이터이다. 이 데이터파일을 열어보면 행에는 DNA유전자 배열이 있고 열에는 해당 DAN구조 칩에 대한 sample들이 존재한다.
데이터 전처리 과정에서는 데이터를 정리하고 확인하는 작업을 진행한다. 우선적으로 데이터 셋자체를 보고 변수의 목록을 확인한다. 그 후 집중하고자하는 데이터 변수를 결정해야한다. 그 후에는 결측치와 이상치를 제거하거나 대체하는 방식을 진행해야한다. 이렇게 진행되는 것을 탐색적 데이터 분석(EDA, Exploratory Data Analysis) 라고 한다. 하지만 일반적으로 column에는 65,000개의 정보가 들어가고 row에는 100만개의 데이터가 들어가게 된다. 이때 유전자 정보를 변수로 쓰기때문에 column에 적는 경우가 많은데 유전자 정보는 정리하면 32만개 정도가 된다. 이러한 문제를 해결하기 위해서 우선 row에 입력하고 행렬데이터를 transpose(전치)시키게된다. - DNA배열을 다루기 때문에 핵심적인 데이터는 문자의 형태를 띄게 된다. 이때 stringsAsFactors를 사용해야 Factor요소로 들어오게 될 때 발생하는 문제를 막을 수 있다. 가져온 데이터의 앞 부분 데이터를 보고 제대로 가져왔는 지 확인을하고
dim()을 통해 전체 데이터 수가 알맞게 가져와졌는지도 확인한다. - 앞서 말했던 방법처럼 이제 원하는 데이터를 형식에 알맞게 기록해야한다. column입력 갯수에 맞춰서 Transpose Matrix(전치 행렬)를 만들어야한다. 그렇기 때문에
t(as.matrix(raw_dat[,-1]))을 해준다. 이때 첫번째 column을 제외하고 만드는 이유는 1번 열은 유전자데이터의 이름들이 있기 때문이다. 이 부분은 앞서 말했듯이 string type이기 때문에 따로 관리를 해준다. - sample의 이름을 굳이 관리한다기 보다는 보기 쉽게 s로 이름을 만들어준다. 그리고 다시 열이름은 원래 raw_dat의 1열 이름들의 형식을 입력해주면 된다.
이로써 데이터 전처리 과정이 끝났다.
- csv파일로 저장된 Ex_data.csv파일은 Lung Cancer Microarray data 이다. 특정 유전자의 발현량이 폐암에 영향을 미치는 가에 대해 분석할 때 사용하는 데이터이다. 이 데이터파일을 열어보면 행에는 DNA유전자 배열이 있고 열에는 해당 DAN구조 칩에 대한 sample들이 존재한다.
결측치 처리 (Missing Value Handling)
- Missing Value Handling
1 2 3 4 5 6 7 8
indx <- which(is.na(dat_mat),T) indx col_ind = indx[,2] col_m = apply(dat_mat[,col_ind],2, mean, na.rm = T) col_m dat_mat[indx] = col_m sum(is.na(dat_mat)) dim(dat_mat)
이제 데이터를 정제하는 과정인 전처리 과정이 끝났으니 결측치를 처리해줘야한다. 결측치 처리가 중요한 이유는 데이터 분석에서 자주 나오는 단어인 outlier(이상치)들이 생각보다 데이터 분석에서 많은 문제를 일으킨다. outlier들은 데이터의 전체적인 구조를 변화시키는 결과를 일으킬 수도 있다. 보통 결측치인 NA들은 특정 값으로 대체를 하게 되는데 이 과정에서 결측치와 이상치들을 잘못 만질경우 전체 데이터에 손상이 일어날 수 있다. 이러한 이유로 결측치와 이상치를 처리를 잘 해줘야한다.
- 우선
which()명령어와is.na()를 활용해서 결측치의 인덱스를 알아낸다. 이렇게 알아낸 정보들은 결측치가 어디에 있는지를 갖고 있는 데이터가 된다. 이후apply()를 활용해서 결측치가 존재하는 인덱스의 값은 그 열의 평균으로 값을 변경해준다. 이를 평균대치법(Mean Imputation)이라고 한다. 이후sum(is.na())로 전체 데이터에 존재하는 결측값의 수를 확인해본다.
- 우선
데이터 전처리 2 (Data Cleaning 2)
- Data Cleaning 2
1 2 3 4 5 6 7 8 9 10 11 12
uq_names <- unique(colnames(dat_mat)) p <- length(uq_names); n <- dim(dat_mat)[1] expr_dat <- matrix(0,n,p) for (i in 1:p) { expr_dat[,i] = apply(as.matrix(dat_mat[,colnames(dat_mat) == uq_names[i]]), 1,mean) cat("\n",i,"-th step") } colnames(expr_dat) <- uq_names rownames(expr_dat) <- rownames(dat_mat) head(expr_dat[,1:20]) dim(expr_dat); sum(is.na(expr_dat))
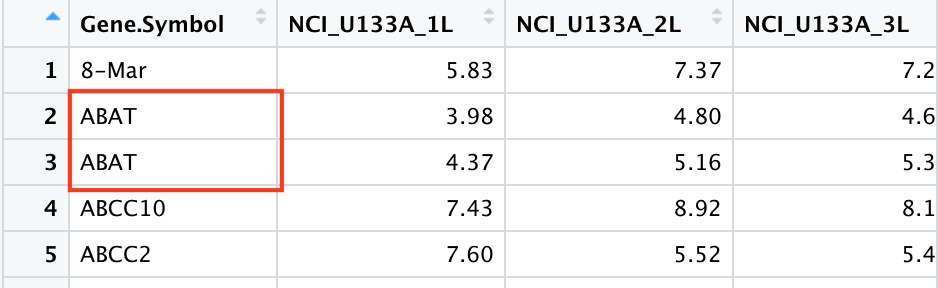
- 가져온 데이터를 잘 살펴보면 중복데이터들이 존재한다.
![R9_d1]()
이런 중복데이터도 처리를 해줄 필요가 있다. 두 데이터의 평균으로 값을 대표해서 적어준다. 이때 이러한 데이터를 처리할 때는 idea가 중요하다.- unique한 유전자인가?
- unique한 유전자의 갯수만큼 평균값 계산을 해준다.
- 선택된 유전자가 원래 데이터에서 어떤 위치에 있는지 확인한다.
unique()를 사용하면 데이터에서 중복된 값을 처리해줄 수 있다. 이렇게 중복된 이름을 우선 제거하고 uq_names에 저장해준다. (1번 과정)- expr_dat라는 임시 데이터를 원본 데이터 크기만큼 만들어준다.
- 그 후 for문을 사용해서 expr_dat에 원본 데이터 값을 갖는 데이터들의 평균치로 채워준다. 이 과정에서 열의 값을 확인한다. uq_names의 i번째 인덱스 값과 원본 데이터의 열의 i번째 값이 같은 부분들을 확인해주는 과정을 진행한다. (2, 3번 과정)
이렇게 기본적인 데이터 전처리와 결측치 대치 과정이 끝났다.
- 가져온 데이터를 잘 살펴보면 중복데이터들이 존재한다.
탐색적 데이터 분석 (EDA, Exploratory Data Analysis)
- EDA
- EDA는 위에서 진행한 데이터 분석 과정을 일컫는다. 데이터 구조와 특징을 우선적으로 분석하고 분석한 결과를 통해 데이터 변환, 축소등의 데이터 손질을 하는 데이터 분석기법이다.
- 일반적으로 EDA 기법은 2가지 정도가 있다.
- 그래프를 이용한 탐색 (Search by Graph)
- 다양한 그래프로 데이터를 요약하고 특징을 관리할 수 있다.
- 통계표 및 통계량을 통한 탐색 (Search by Statistics data)
- 평균, 분산, 분위수 등의 분포 파악을 위한 모수 추정값을 요약한다.
- 그래프를 이용한 탐색 (Search by Graph)
데이터 결측값과 특이값 (Missing Value & Outlier)
- Missing Value & Outlier
결측값(Missing Value) 이란 데이터 수집과정에서 수집이 되지않았거나 손실이 발생을 해서 데이터 일부가 누락되어 측정된 것을 말한다. 이런 결측치를 대치하는 방법도 다양하게 존재한다.
특이값(Outlier) 은 일상적으로 접하는 데이터에서 대부분의 데이터가 따르는 경향에서 크게 벗어나 있는 데이터를 말한다. 특이값은 분석 모형 결과에 크게 영향을 줄 수 있기 때문에 전처리 과정에서 식별해서 모형에서는 제외한다. 특이값이 발생하는 것이 사실 이상한 것은 아니다. 중요한 것을 왜 특이값(Outlier)이 발생했는가? 이다. 특이값이 발생한 이유에 따라서 해당 특이값이 유의미한지, 무의미한지를 결정해주기 때문이다.
목요일
막대그래프 (barplot)
- barplot
1 2 3 4 5 6 7 8 9 10 11 12
dat = read.table(file="Ex211_tab.txt", header = T, sep = '\t', fileEncoding = "CP949",encoding = "UTF-8") head(dat) dat$Job attach(dat);Job search() quartz() plot(Job, main="직업의 막대그림", ylab="인원수(명)",ylim=c(0,15), xlim=c(0,5)) box() freq = table(dat[,6]) barplot(freq) detach(Job)
데이터의 특정 변수 값을 $연산자를 통해 직접적으로 컨트롤할 수 있는 권한을 얻을 수 있다. 이런 번거로움을 덜기 위해서
attach()를 활용하면 원하는 데이터 셋의 변수에 변수이름만으로 직접 접근을 할 수 있다. 활용 후에detach()로 원래대로 돌려주는 것이 좋다. quartz()나 x11()을 활용하면 별도의 디스플레이에 그래프를 띄울 수 있다. 나는 맥북이라 x11()을 사용하기 위해서 quartz()를 쓰므로 quartz로 명령어를 쓴다. 일반적으로는 x11()사용해도 괜찮다.plot()을 쓸때 어떤 값을 활용할 지 정하고 제목과 x,y축의 이름을 정해줄 수 있다. 이때ylim과xlim을 사용하면 x,y축의 최대 / 최소 한계 값을 지정해줄 수 있다. plot은 일반적인 그래프로 나타내준다.barplot()을 쓰면 막대그래프 형식으로 나타난다. 이때 내부에 들어갈 값은 빈도수를 확인할 수 있는 값이어야한다.
barplot2
- barplot2
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18
data("VADeaths") install.packages("gplots") library(gplots) x11() barplot2(t(VADeaths), beside = TRUE, col = gray(seq(0.4,0.9,length=5)), legend=colnames(VADeaths), ylim = c(0,100)) title(main = "Death Rates in Virginia", font.main=4) hh <- t(VADeaths)[,5:1] mybarcol <- "gray20" cil <- hh * 0.85 ciu <- hh * 1.15 mp <- barplot2(hh, beside = T, col = gray(seq(0.4,0.9,length = 5)), legend = colnames(VADeaths), ylim = c(0,100), main = "Death Rates in Virginia", font.main = 4, sub = "Faked 95 percent error bars", cex.names = 1.5, plot.ci = T, ci.l = cil,ci.u = ciu,plot.grid = T) box()
barplot2 실험데이터 분석을 할 때 쓰기 좋은 함수이다. “gplots”패키지에 들어있으며 패키지 설치 진행후 사용을 해야한다.
- barplot2에서 beside가 T인 경우는 일반적은 막대그래프의 형태이다. F인 경우에는 하나의 막대그래프에 누적형태로 나타나게 된다. legend는 막대하나하나에 어떤 의미가 있는지를 보여주는 형식을 출력해준다.
- plot에 신뢰구간도 보여줄 수 있다. 내부 parameter에 plot.ci는 plot confidence interval의 약자이다. 즉 신뢰구간 설정 여부를 말해준다. 이 값을 TRUE로 해준 경우 ci.l과 ci.u 값들을 설정해줘야한다.
원 그래프 (pie chart)
- pie chart
1 2 3 4 5 6
pie(freq, main = "직업의 원그림") pie(rep(1,24), col = rainbow(24), radisu = 0.9) pie.sales <- c(0.12,0.3,0.26,0.16,0.04,0.12) lbl = c("Blueberry","Cherry","Apple","Boston Cream", "Other","Vanilla Cream") names(pie.sales) = paste0(lbl, " (", pie.sales*100, "%)") pie(pie.sales, col = rainbow(length(pie.sales)))
일반적인 원형그래프를 보여준다. frequency를 당연하게 입력해줘야한다., 해당 비율만큼 알아서 원 그래프의 면적을 채워준다. names를 쓰면 원 그래프의 항목 이름을 설정해줄 수 있다.
히스토그램 (histogram)
- histogram
1 2 3 4
x <- expr_dat[,10] x11() hist(x, breaks = 20, col = "gray", main = uq_names[10]) hist(x, breaks = 20, freq = F ,col = "lightblue", main = uq_names[10])
일반적인 히스토그램을 출력해준다. breaks는 상자의 갯수를 지정해준다. 이때 frequency는 default가 T인데, F로 설정할 경우 히스토그램을 밀도값으로 보여준다. 데이터의 갯수가 아니라 해당 데이터가 차지하는 밀도로 보여준다. 일종의 상대도수그래프와 같다.
- hist함수의 plot을 F로 변경해주면 그래프는 그려지지 않고 그래프를 그릴 때 필요한 데이터들을 모두 보여준다.
Comments powered by Disqus.